Nurcan Tuncbag, PhD

Postdoctoral Associate
Massachusetts Institute of Technology
Department of Biological Engineering
Contact information
77 Massachusetts Avenue
Building 16 Room 244
Phone: 617-756-4411
[email protected]
Biography
I received a B.S. degree from Istanbul Technical University, Turkey (Chemical Engineering) and a Master’s (Computational Sciences and Engineering) and Ph.D. (Computational Sciences and Engineering) from Koc University, Turkey (advisors: Prof Ozlem Keskin and Prof. Attila Gursoy). During my Ph.D, I closely collaborated with Prof. Ruth Nussinov from National Cancer Institute (NCI). My Ph.D. work focused on structural modeling of protein interactions and incorporating them into the protein interaction networks. We developed an algorithm for large-scale prediction of protein-protein interactions and assembly of protein complex structures which integrates structural and evolutionary similarity with flexible refinement and energy calculations. Then, I joined Ernest Fraenkel’s group at Massachusetts Institute of Technology as a Postdoctoral Associate and began working on revealing how the networks of interactions among proteins and genome are altered in cells during disease.
>> My CV etc. are available at http://www.mit.edu/~ntuncbag/
Massachusetts Institute of Technology
Department of Biological Engineering
Contact information
77 Massachusetts Avenue
Building 16 Room 244
Phone: 617-756-4411
[email protected]
Biography
I received a B.S. degree from Istanbul Technical University, Turkey (Chemical Engineering) and a Master’s (Computational Sciences and Engineering) and Ph.D. (Computational Sciences and Engineering) from Koc University, Turkey (advisors: Prof Ozlem Keskin and Prof. Attila Gursoy). During my Ph.D, I closely collaborated with Prof. Ruth Nussinov from National Cancer Institute (NCI). My Ph.D. work focused on structural modeling of protein interactions and incorporating them into the protein interaction networks. We developed an algorithm for large-scale prediction of protein-protein interactions and assembly of protein complex structures which integrates structural and evolutionary similarity with flexible refinement and energy calculations. Then, I joined Ernest Fraenkel’s group at Massachusetts Institute of Technology as a Postdoctoral Associate and began working on revealing how the networks of interactions among proteins and genome are altered in cells during disease.
>> My CV etc. are available at http://www.mit.edu/~ntuncbag/
Simultaneous reconstruction of multiple signaling pathways
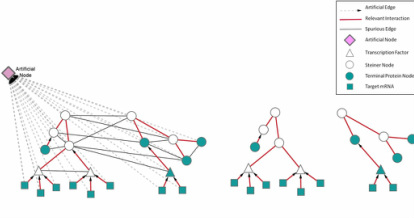
Signaling and regulatory networks are essential for cells to control processes such as growth, differentiation and response to stimuli. Although a variety of “omic” data sources are available to probe signaling pathways, these data are typically sparse and noisy. My work has been focused on formulating an approach for using “omic” data to reconstruct multiple pathways that are altered in a particular condition. Previously, we have shown that these data could be efficiently analyzed by solving the prize collecting Steiner tree (PCST) problem to reveal a biologically relevant signaling pathway composed of a subset of the detected proteins through other undetected proteins present in the interactome. I modified this approach to simultaneously discover multiple pathways searching for “forests” consisting of multiple trees, and using directed edges in the interactome. I used the approach to examine changes in signaling that occur in glioblastoma multiforme (GBM). This approach discovered both overlapping and independent signaling pathways. Although the algorithm was not provided with any information about the phosphorylation status of cell surface receptors, it identifies a small set of clinically relevant receptors among hundreds present in the interactome. The method can be applied efficiently to “omic” data and is able to reconstruct networks that are enriched in functionally and clinically relevant proteins. Currently, I am working on incorporating existing pharmaceutical or biological agents into this algorithm to suggest possible combined therapy to treat specific diseases.
PRISM: Fast and Accurate Modeling of Protein-Protein Interactions Using Template-Based
Flexible Docking

Prediction of protein-protein interactions at the structural level on the proteome scale is important because it allows prediction of protein function, helps drug discovery and takes steps toward genome-wide structural systems biology. We provide a protocol (termed PRISM, protein interactions by structural matching) for large-scale prediction of protein-protein interactions and assembly of protein complex structures. The method consists of two components: rigid-body structural comparisons of target proteins to known template protein-protein interfaces and flexible refinement using a docking energy function. The PRISM rationale follows our observation that globally different protein structures can interact via similar architectural motifs. PRISM predicts binding residues by using structural similarity and evolutionary conservation of putative binding residue 'hot spots'. Ultimately, PRISM could help to construct cellular pathways and functional, proteome-scale annotation. PRISM is implemented in Python and runs in a UNIX environment. The program accepts Protein Data Bank-formatted protein structures and is available at http://prism.ccbb.ku.edu.tr/prism_protocol/.
>> For details: http://www.ncbi.nlm.nih.gov/pubmed/21886100
>> For details: http://www.ncbi.nlm.nih.gov/pubmed/21886100
A Review on Structure-Based Modeling of Protein-Protein Interactions
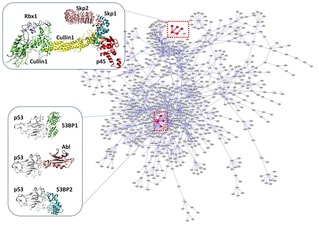
The vast majority of the chores in the living cell involve protein-protein interactions. Providing details of protein interactions at the residue level and incorporating them into protein interaction networks are crucial toward the elucidation of a dynamic picture of cells. Despite the rapid increase in the number of structurally known protein complexes, we are still far away from a complete network. Given experimental limitations, computational modeling of protein interactions is a prerequisite to proceed on the way to complete structural networks. In this work, we focus on the question 'how do proteins interact?' rather than 'which proteins interact?' and we review structure-based protein-protein interaction prediction approaches. As a sample approach for modeling protein interactions, PRISM is detailed which combines structural similarity and evolutionary conservation in protein interfaces to infer structures of complexes in the protein interaction network. This will ultimately help us to understand the role of protein interfaces in predicting bound conformations.
>> For details: http://www.ncbi.nlm.nih.gov/pubmed/21572173
>> For details: http://www.ncbi.nlm.nih.gov/pubmed/21572173
Learning-based Prediction of Binding Hot Spots in Protein-Protein Interfaces & Hotpoint Webserver
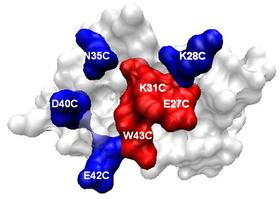
The energy distribution along the protein-protein interface is not homogenous; certain residues contribute more to the binding free energy, called 'hot spots'. Here, we present a web server, HotPoint, which predicts hot spots in protein interfaces using an empirical model. The empirical model incorporates a few simple rules consisting of occlusion from solvent and total knowledge-based pair potentials of residues. The prediction model is computationally efficient and achieves high accuracy of 70%. The input to the HotPoint server is a protein complex and two chain identifiers that form an interface. The server provides the hot spot prediction results, a table of residue properties and an interactive 3D visualization of the complex with hot spots highlighted. Results are also downloadable as text files. This web server can be used for analysis of any protein-protein interface which can be utilized by researchers working on binding sites characterization and rational design of small molecules for protein interactions. HotPoint is accessible at http://prism.ccbb.ku.edu.tr/hotpoint.
>> For details: http://www.ncbi.nlm.nih.gov/pubmed/21572173 http://www.ncbi.nlm.nih.gov/pubmed/19357097
>> For details: http://www.ncbi.nlm.nih.gov/pubmed/21572173 http://www.ncbi.nlm.nih.gov/pubmed/19357097
A Rigorous Analysis of Protein Data Bank - Architectures and Functional Coverage of Protein Interfaces
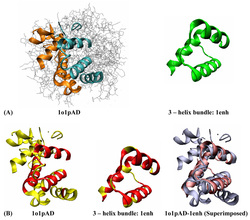
The diverse range of cellular functions is performed by a limited number of protein folds existing in nature. One may similarly expect that cellular functional diversity would be covered by a limited number of protein-protein interface architectures. Here, we present 8205 interface clusters, each representing a unique interface architecture. This data set of protein-protein interfaces is analyzed and compared with older data sets. We observe that the number of both biological and crystal interfaces increases significantly compared to the number of Protein Data Bank entries. Furthermore, we find that the number of distinct interface architectures grows at a much faster rate than the number of folds and is yet to level off. We further analyze the growth trend of the functional coverage by constructing functional interaction networks from interfaces. The functional coverage is also found to steadily increase. Interestingly, we also observe that despite the diversity of interface architectures, some are more favorable and frequently used, and of particular interest, are the ones that are also preferred in single chains.
>> For details: http://www.ncbi.nlm.nih.gov/pubmed/18620705
>> For details: http://www.ncbi.nlm.nih.gov/pubmed/18620705
